人基因組重測(cè)序
人基因組重測(cè)序可全面掃描人類基因組上的變異信息,一次性挖掘大量的生物標(biāo)記物,該技術(shù)準(zhǔn)確性高、可重復(fù)性好、定位精確,已被廣泛應(yīng)用于疾病、癌癥的基因組研究。
人全基因組重測(cè)序(二代測(cè)序/三代測(cè)序)
人全基因組重測(cè)序(Whole-Genome Sequencing,WGS)對(duì)人類不同個(gè)體或群體進(jìn)行基因組測(cè)序,在全基因組范圍內(nèi)挖掘DNA水平遺傳變異,全面挖掘基因組的SNV/InDel/CNV/SV等各類變異。其中,三代測(cè)序憑借超長(zhǎng)讀長(zhǎng)、無(wú)偏向性等優(yōu)勢(shì)對(duì)大片段的結(jié)構(gòu)變異檢測(cè)有更好的檢出率,同時(shí)對(duì)一些復(fù)雜的SV也能夠很好的檢測(cè)出來(lái)。人全基因組重測(cè)序是目前獲得人基因信息較為全面的測(cè)序手段,也是研究與輔助治療人類疾病重要的方式。
人全外顯子組測(cè)序
全外顯子組測(cè)序(Whole Exome Sequencing,WES)是利用探針捕獲和富集外顯子區(qū)域DNA序列,再通過(guò)高通量測(cè)序和分析獲得與疾病相關(guān)的致病突變信息。人類基因組外顯子只占整個(gè)基因組的1%,但其包含了約85%的致病突變,WES測(cè)序深度更高,有利于發(fā)現(xiàn)常見(jiàn)變異、低頻變異和罕見(jiàn)變異,且測(cè)序費(fèi)用更低、周期更短。
人全外顯子關(guān)聯(lián)分析
通過(guò)全外顯子組測(cè)序(WES)測(cè)序技術(shù)檢測(cè)患者之間的基因差異,找出導(dǎo)致致病性差異的基因變異,探討基因型差異與表型差異之間的相關(guān)性,以期預(yù)測(cè)疾病表型發(fā)生,使治療更加個(gè)體化。
低深度WGS CNV-seq
CNV(Copy Number Variation)是一種常見(jiàn)的基因組變異,研究人基因組中的 CNV有助于發(fā)現(xiàn)新的遺傳致病因子和了解其多態(tài)性水平與進(jìn)化歷程。利用低深度 WGS 檢測(cè)人基因組 CNV 具有分辨率高,檢測(cè)全面且成本低廉等優(yōu)勢(shì),適用于大規(guī)模樣本的 CNV 分析。
應(yīng)用介紹
- 個(gè)體差異和種群遺傳學(xué):研究個(gè)體之間的DNA差異,有助于解析人類遺傳變異信息,以及變異如何影響個(gè)體的健康和易感性;
- 疾病發(fā)生機(jī)制:比較正常和疾病狀態(tài)下的DNA差異,挖掘與疾病發(fā)生和進(jìn)展相關(guān)的變異,增進(jìn)對(duì)疾病發(fā)生和發(fā)展過(guò)程的理解;
- 腫瘤進(jìn)化機(jī)制:利用更深的基因覆蓋,挖掘更全面的腫瘤變異信息,解析腫瘤致病機(jī)理、轉(zhuǎn)移/復(fù)發(fā)機(jī)制、易感基因、耐藥機(jī)制、異質(zhì)化及腫瘤進(jìn)化;
- 藥物基因組:尋找藥物療效/安全性相關(guān)遺傳突變、研究遺傳變異在個(gè)體藥物反應(yīng)差異性中發(fā)揮的作用,指導(dǎo)精準(zhǔn)用藥及個(gè)體化治療;
- 基因診斷:幫助提高臨床上難以診斷疾病的診斷率;
文章案例
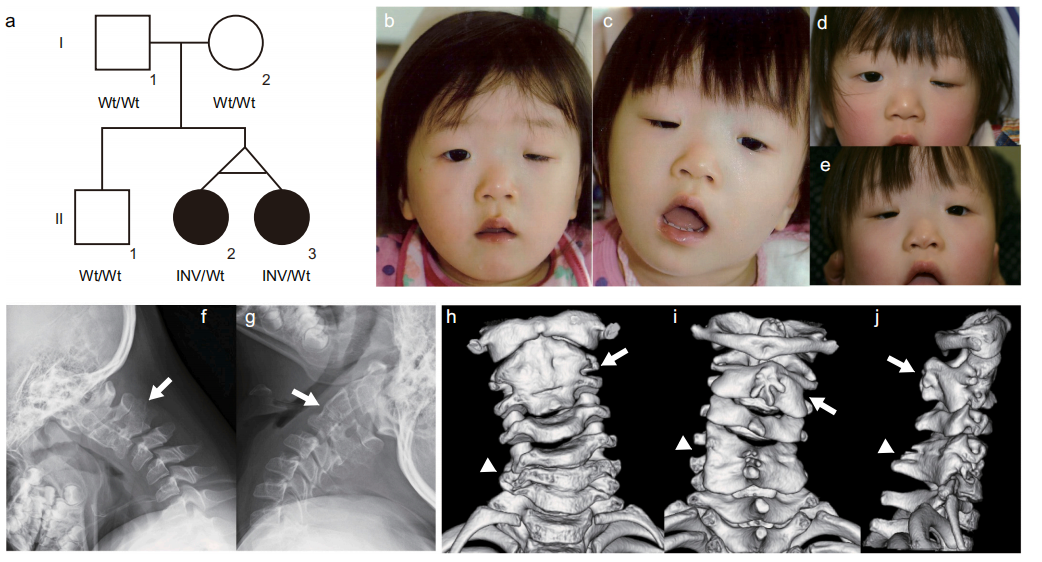
致病性SV及樣本情況
HiFi測(cè)序揭示綜合征性智力障礙的致病性結(jié)構(gòu)變異
日本橫濱市立大學(xué)醫(yī)學(xué)院和大阪市婦女兒童醫(yī)院的科學(xué)家對(duì)一對(duì)患有綜合征智力障礙的同卵雙胞胎女孩進(jìn)行PacBio HiFi測(cè)序,以探索之前未被全外顯子測(cè)序檢測(cè)到的致病變異。通過(guò)Trio家系樣本和數(shù)據(jù)庫(kù)過(guò)濾,獲得一個(gè)12kb的倒位變異,該倒位直接破壞了CPNE9和BRPF1兩個(gè)基因并可能導(dǎo)致基因功能喪失。此外,基于Trio樣本的單倍型定相分析還發(fā)現(xiàn)該倒位遺傳自母親。該研究清楚地表明了亞顯微拷貝中性倒位的重要性, 以及利用長(zhǎng)讀長(zhǎng)測(cè)序技術(shù)檢測(cè)遺傳病中這種變異的價(jià)值。
參考文獻(xiàn):Mizuguchi Takeshi, Okamoto Nobuhiko, Yanagihara Keiko et al. Pathogenic 12-kb copy-neutral inversion in syndromic intellectual disability identified by high-fidelity long-read sequencing[J]. Genomics, 2021, 113: 1044-1053.
北京市昌平區(qū)科技園區(qū)生命園路4號(hào)院5號(hào)樓
客服熱線:400-610-8005

